ONGOING RESEARCH FOR BETTER EVIDENCE
At Abbott, we are committed to investing in clinical trials and evidence to continue building better solutions for patients worldwide and to build trust in the safety and performance of our portfolio of life-changing structural heart devices. With over 45 planned clinical trials, we take great pride in building confidence for physicians and patients.
This page is intended to provide clinical trial information only. The products and procedures discussed on this page are currently in clinical study and are not approved
for commercial use.
STRUCTURAL INTERVENTIONS


1. The Amulet delivery system is introduced via femoral venous puncture and enters the right atrium.

2. A transseptal puncture allows the Amulet delivery system to enter the left atrium.

3. The Amulet occluder is guided to the left atrial appendage using echocardiography and fluoroscopy. Retention wires on the lobe secure the occluder, while the disc expands to seal the appendage orifice.
ADVANCE LAA is a prospective, multi-center observational study intended to characterize real-world outcomes of patients receiving the commercially available Amplatzer™ Amulet™ LAA Occluder in the United States.
- Procedural overview
The Amulet occluder is delivered in a minimally invasive transcatheter approach through a femoral venous puncture. The Amulet occluder is introduced to the left atrium via an interatrial transseptal puncture. The lobe secures the Amulet occluder within the left atrial appendage, while the disc completely seals the appendage at the orifice.
- Eligibility criteria*
Key Inclusion Criteria
- Intended for LAAO with the Amulet device
- At least 18 years of age
- Willing and capable of providing informed consent and participating in all testing associated with this clinical study
Key Exclusion Criteria- Presence of other conditions that, in the investigator's opinion, could limit the subject's ability to participate in follow-up
*Additional inclusion and exclusion criteria as listed on clinicaltrials.gov
- Trial design
The ADVANCE LAA study will enroll approximately 1000 subjects at up to 50 US clinical sites. Subjects will be followed through 24 months in accordance with each site’s standard care practices.
- Clinical trial endpoints
- Composite of all-cause death, ischemic stroke, systemic embolism, or device/procedure related complications requiring an invasive surgical or percutaneous intervention within 7 days of implant or hospital discharge, whichever is later
- Device closure (defined as residual jet around the device ≤ 3mm) at the first follow-up visit documented by trans-esophageal echocardiography (TEE), as assessed by an independent core laboratory
- Composite of ischemic stroke or systemic embolism through 24 months
To learn more about the ADVANCE LAA Trial, including eligibility criteria and study locations, please visit the ClinicalTrials.gov website.

For people with atrial fibrillation (AFib), irregular electrical impulses in the upper chambers can cause an increased risk for developing blood clots, which can lead to a stroke. The left atrial appendage can be a major source of blood clots in patients with AFib. The CATALYST clinical trial will evaluate the safety and effectiveness of the Amplatzer™ Amulet™ Left Atrial Appendage Occluder compared to the treatment of blood thinner medication in patients with non-valvular AFib who are at an increased risk for stroke and who are recommended for long-term blood thinner medication.
CAUTION: The Amplatzer Amulet device (for CATALYST indications) is an investigational device. Limited by Federal (U.S.) law to investigational use only.
1. The Amulet delivery system is introduced via femoral venous puncture and enters the right atrium.
2. A transseptal puncture allows the Amulet delivery system to enter the left atrium.
3. The Amulet Occluder is guided to the left atrial appendage using echocardiography and fluoroscopy. Retention wires on the lobe secure the occluder, while the disc expands to seal the appendage orifice.
- Procedural overview
The Amulet occluder is delivered in a minimally invasive transcatheter approach through a femoral venous puncture. The Amulet occluder is introduced to the left atrium via an interatrial transseptal puncture. The lobe secures the Amulet occluder within the left atrial appendage, while the disc completely seals the appendage at the orifice.
- Eligibility criteria*
Key Inclusion Criteria
- Documented paroxysmal, persistent, or permanent non-valvular AF (documentation must include an electrocardiogram, Holter, or event recorder)
- At high risk of stroke or systemic embolism, defined as a CHA2DS2-VASc score of ≥ 3 for women and ≥ 2 for men
- Eligible for long-term NOAC therapy
Key Exclusion Criteria- Requires long-term OAC therapy for a condition other than AF
- Known contraindication to, or allergic to, aspirin, clopidogrel, or OAC medication use
- Has undergone atrial septal defect (ASD) repair or has an ASD closure device present
- Has undergone patent foramen ovale (PFO) repair or has a PFO closure device implanted
*Additional inclusion and exclusion criteria as listed on ClinicalTrials.gov - Trial design
The CATALYST Trial is a prospective, randomized, multicenter active control worldwide trial. Subjects are randomized in a 50/50 ratio between the Amulet LAA occlusion device and a commercially available NOAC blood thinner medication.
- Clinical trial endpoints
- Composite of ischemic stroke, systemic embolism, or CV mortality
- Major bleeding or clinically relevant non-major bleeding (CRNMB) events, excluding procedure related events
- Composite of ischemic stroke or systemic embolism
Find a clinical trial site near me
The PARADIGM trial for patients with clinically significant paravalvular leak (PVL) who are eligible for transcatheter closure is now underway and will be studying approximately 200 subjects in up to 25 medical centers worldwide. The trial is a prospective, multicenter, single arm study to demonstrate the safety and effectiveness of the AVP III as a treatment for clinically significant PVLs following surgical implant of a mechanical or biological heart valve implanted in the aortic or mitral position.
CAUTION: Investigational device. Limited by federal (U.S.) law to investigational use only.
- Procedural overview
After surgical heart valve replacement procedure, separation between the new valve and the existing heart tissue can occur, creating paravalvular leak (PVL). Historically, repair of PVL required another invasive surgery to close the leak. In PARADIGM, patients will receive the Amplatzer Valvular Plug III (AVP III), delivered through a minimally invasive transcatheter approach. The AVP III is deployed at the origin of the PVL , and the unique geometry of the AVP III allows for sealing the area between the artificial valve and native tissue.



- Eligibility criteria*
Key Inclusion Criteria
- US & CA: Patients with a mechanical or bioprosthetic surgical aortic or mitral valve
- EU: Patients with a mechanical surgical aortic or mitral valve
- Patients with significant PVL, showing symptoms of heart failure or hemolytic anemia
Key Exclusion Criteria- PVL originates from transcatheter aortic or mitral valve replacement (TAVR/TMVR)
- Rocking valve motion or extreme dehiscence of valve from native tissue
- Patients with multiple clinical PVLs or dual aortic and mitral PVLs
*Additional inclusion and exclusion criteria as listed on ClinicalTrials.gov - Trial design
The PARADIGM study has a prospective, single-arm, multicenter design in which 200 subjects, meeting all eligibility requirements and undergoing a transcatheter AVP III implant or implant attempt, will be followed for 12 months post-implant.
- Clinical trial endpoints
The primary endpoint is a composite of safety and effectiveness measures evaluating the percent of patients successfully treated with an AVP III for the reduction of PVL, where success is defined as:
- Transcatheter placement of the AVP III in the intended location without interfering with the surgical valve function on exit from procedure
- Reduction in PVL by greater than or equal to two grades on exit from procedure
- Freedom from intra-procedural death
- Freedom from unplanned surgical procedure or transcatheter reintervention related to the AVP III through 30 days
To learn more about the PARADIGM Trial, including eligibility criteria and study locations, please visit the ClinicalTrials.gov website.
TRANSCATHETER SOLUTIONS
CEPHEA EFS | CEPHEA MVD REGISTRY | ENVISION TRIAL | REPAIR MR TRIAL | SUMMIT TRIAL | TRILUMINATE TRIAL
Cephea Early Feasibility Study (EFS) evaluates transcatheter mitral valve replacement (TMVR) with the Cephea™ Mitral Valve System.
Cephea EFS is now underway and will treat up to 50 patients at up to 20 sites globally.
The objective of Cephea EFS is to evaluate the preliminary safety and effectiveness of the Cephea™ Mitral Valve System for the treatment of symptomatic patients with mitral valve disease (including mitral regurgitation, mitral stenosis and mixed mitral valve disease) in whom transcatheter therapy is deemed more appropriate than open heart surgery.
CAUTION: investigational device. Limited by federal (U.S.) Law to investigational use only.
- Procedural overview
The Cephea™ Mitral Valve System utilizes transfemoral access and a transseptal approach for the delivery and implantation of a radially symmetric self-expanding mitral valve.

Pre-deployment

Deployment

Valve Deployed
- Eligibility criteria
Key Inclusion Criteria
- MVD resulting in mitral regurgitation (MR ≥ Grade III) and/or severe mitral stenosis
- NYHA Functional Classification ≥ II (if Class IV, subject must be ambulatory)
- The local site heart team determines that the subject has been adequately treated per applicable standards and is better suited to TMVR over surgery
- Left ventricular ejection fraction ≥ 30%
- Left ventricular end diastolic diameter ≤ 70mm
Key Exclusion Criteria
- Prior surgical or interventional treatment that interferes with Cephea valve delivery or function.
- Any planned cardiac surgery within the next 12 months.
- ACC/AHA Stage D heart failure or prior orthotopic heart transplantation.
- Severe tricuspid regurgitation, tricuspid valve disease, or aortic valve disease requiring surgery or transcatheter intervention.
- Evidence of right-sided congestive heart failure or significant right ventricular dysfunction.
- Subject is undergoing hemodialysis due to chronic renal failure.
- Subjects with comorbidities that are likely to result in a life expectancy of less than 12 months
- The presence of other anatomic or comorbid conditions, or other medical, social, or psychological conditions that, in the Investigator’s opinion, could limit the subject’s ability to participate in the clinical investigation or to comply with follow-up requirements, or impact the scientific soundness of the clinical investigation results.
- Trial design
Cephea EFS is a multi-center, single-arm, prospective, interventional study.
Subjects who sign consent will be assessed for implant and study suitability through an internal Sponsor review and a Physician Review Team review. Subjects who pass both reviews are approved for device implant.
Enrolled and implanted subjects will be followed prior to discharge, and at 30 days, 3 months, 6 months, 12 months and annually through 5 years.

- Clinical trial endpoints
Two primary endpoints will be used to characterize the safety and effectiveness of the Cephea™ Mitral Valve System and will be measured at 30 days post-implant:
- Safety: Freedom from all-cause mortality
- Effectiveness: MR ≤ Grade I as classified by the Echocardiography Core Lab.
Additional data will be collected to measure device safety and effectiveness, including:
- MVARC-defined Technical, Device, Procedural and Patient Success
- Annualized rate of heart failure hospitalization
- Functional measurements
- Quality of life measurements.
To learn more about the Cephea Early Feasibility Study, including eligibility criteria and study locations, please visit the ClinicalTrials.gov website.
Cephea Mitral Valve Disease (MVD) Registry is designed to collect data on clinical outcomes of symptomatic MVD patients who may be candidates for Transcatheter Mitral Valve Replacement (TMVR).
Despite a number of treatments options available for MVD, many patients are not candidates for these treatments. TMVR is a promising option for these patients. The MVD Registry focuses on following patients whose MVD symptoms continue despite undergoing optimal medical therapy, and for whom TMVR is considered a better option than open-heart surgery or other mitral valve repair procedures.
The objective of this Registry is:
- To better understand how MVD progresses and how different treatments impact patient symptoms and outcomes.
- Collect data on the clinical outcomes of TMVR candidates who, due to anatomic limitations, are unable to undergo TMVR. Understanding these outcomes will be helpful to compare to TMVR treatment studies.
MVD Registry is now underway and will be enrolling up to 1000 patients at up to 100 sites in the globally.
CAUTION: investigational device. Limited by federal (U.S.) Law to investigational use only.
- Procedural overview
MVD Registry is a data collection study and does not involve any interventions or study-specific procedures. Subjects will be followed for two years with data being collected on their MVD progression through visits and procedures conducted as part of their routine care.
- Eligibility criteria
Key Inclusion Criteria
- Symptomatic MVD due to moderate-severe mitral regurgitation (MR) and/or severe mitral stenosis (MS) or both moderate MR and moderate MS
- NYHA Functional Classification ≥ II (if Class IV, subject must be ambulatory)
- The local site heart team determines that the subject has been adequately treated per applicable standards and is better suited to TMVR over surgery or TEER
- Left ventricular ejection fraction ≥ 30%
- Left ventricular end diastolic diameter ≤ 70mm
Key Exclusion Criteria
- Prior surgical or interventional treatment that interferes with Cephea valve delivery or function.
- ACC/AHA Stage D heart failure or prior orthotopic heart transplantation.
- Severe tricuspid regurgitation, tricuspid valve disease, or aortic valve disease requiring surgery or transcatheter intervention.
- Evidence of right-sided congestive heart failure or significant right ventricular dysfunction.
- Subject is undergoing hemodialysis or experiencing chronic renal failure.
- Subjects with comorbidities that are likely to result in a life expectancy of less than 12 months.
- The presence of other anatomic or comorbid conditions, or other medical, social, or psychological conditions that, in the Investigator’s opinion, could limit the subject’s ability to participate in the study or to comply with follow-up requirements.
- Trial design
MVD Registry is a prospective, observational, multi-center study intended to collect data on MVD disease progression, interventions, and clinical outcomes in subjects who may be candidates for TMVR.
Subjects enrolled in the Registry undergo anatomic screening and review by a Physician Review Team (PRT) to confirm they are medically suitable candidates for TMVR. Subjects determined to be medically suitable for TMVR, regardless of their anatomic suitability, will begin follow-up in the Registry. Data will be collected from follow-up visits performed as part of the subject’s routine care at 6, 12, 18 and 24 months.
Subjects enrolled in the Registry who are determined to be both medically and anatomically suitable for TMVR may be consented to undergo further evaluation with the opportunity to be treated under a Cephea interventional clinical study using the Cephea™ Mitral Valve System. If they are treated with the Cephea System, they will be withdrawn from the Registry and will be followed under the interventional study.

- Clinical trial endpoints
MVD Registry is a data collection Registry and therefore there are no pre-defined endpoints. Data collected as part of the MVD Registry may be used for comparison to the endpoints of Cephea interventional clinical investigations as well as for exploratory purposes.
To learn more about the Cephea MVD Registry, including eligibility criteria and study locations, please visit the ClinicalTrials.gov website.
The ENVISION Clinical Trial is studying the Navitor™ Transcatheter Aortic Valve Implantation (TAVI) System in patients with severe, symptomatic aortic stenosis who are deemed low or intermediate surgical risk. Approximately 1,500 patients will be studied at up to 80 medical centers in the United States and up to 15 medical centers outside of the United States.
CAUTION: The Navitor TAVI System (for ENVISION Clinical Trial indications) is an investigational device. Limited by Federal (U.S.) law to investigational use only.
- Procedural overview
Through transfemoral, subclavian or axillary access methods, the Navitor™ Valve will be introduced in the patient’s body using the FlexNav™ Delivery System. The delivery system design facilitates gradual, controlled deployment of the valve. Controlled movement of the sheath will be viewed through cardiac fluoroscopic imaging. Predilate the native aortic valve with an appropriate diameter valvuloplasty balloon. Once the sheath is in position, the Navitor Valve will be delivered and placed within the opened aortic valve (Figure 1). The Navitor Valve is deployed annulus end first from the distal end of the delivery system (Figure 2). If needed, the valve may be resheathed and repositioned up to two times, provided the valve has not been fully deployed. Once the Navitor Valve has been released from its delivery system, it will start functioning. The delivery system will be removed from the patient’s body, and the incision will be closed.
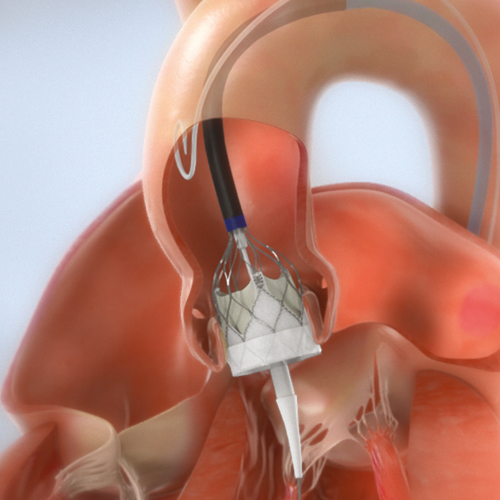
FIGURE 1: Navitor Valve in Aortic Valve
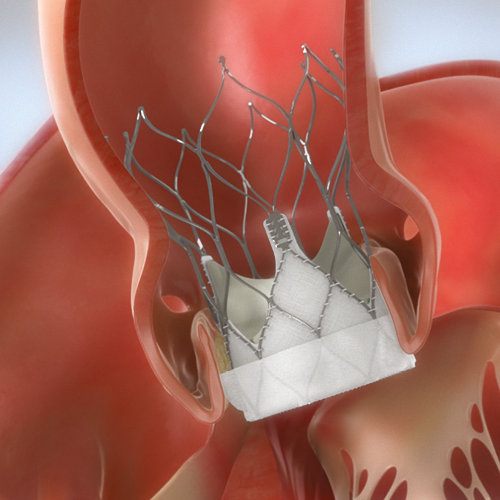
FIGURE 2: Navitor Valve in Position
- Eligibility criteria*
Key inclusion criteria
- Subject is deemed to be at low or intermediate risk for open surgical aortic valve replacement (risk of surgical mortality < 7% at 30 days, considering The Society of Thoracic Surgeons (STS) risk score, overall clinical status, and other clinical comorbidities unmeasured by the risk calculator)
- New York Heart Association (NYHA) Functional Classification of II, III or IV
- Senile degenerative aortic valve stenosis with ECHO-derived criteria, which is defined as aortic valve area (AVA) of ≤ 1.0 cm2 (or indexed effective orifice area (EOA) ≤ 0.6 cm2/m2) and either mean gradient ≥ 40 mmHg or peak jet velocity ≥ 4.0 m/s or Doppler velocity index (DVI) ≤ 0.25
- Aortic annulus diameter of 19–30 mm and ascending aorta diameter of 26–44 mm for the specified valve size listed in the Instructions for Use (IFU), as measured by computerized tomography (CT) conducted within 12 months prior to informed consent
Key exclusion criteria
- Recent (within 6 months prior to index procedure date) cerebrovascular accident (CVA) or transient ischemic attack (TIA)Renal insufficiency (creatinine > 3.0 mg/dL or estimated glomerular filtration rate (eGFR) < 30 mL/min/1.73 m2) and/or end-stage renal disease requiring chronic dialysis
- Need for emergency surgery for any reason
- Hostile chest or conditions or complications from prior surgery that would make the patient be considered high surgical risk
- Significant frailty as determined by the heart team (after objective assessment of frailty parameters) that would indicate high or extreme surgical risk
- Evidence of an acute myocardial infarction within 30 days prior to index procedure
- Mixed aortic valve disease (aortic stenosis and aortic regurgitation with predominant aortic regurgitation 3–4+)
- Aortic valve is a congenital unicuspid or congenital bicuspid valve as verified by echocardiography and computerized tomography
- Severe ventricular dysfunction with left ventricular ejection fraction (LVEF) < 30%, as measured by resting echocardiogram
- Non-calcified aortic valve
*Additional inclusion and exclusion criteria as listed on ClinicalTrials.gov - Trial design
This is a prospective, randomized, controlled, multicenter, investigational clinical study of approximately 1,500 patients at up to 80 medical centers in the United States and up to 15 medical centers outside of the United States. The objective of this clinical investigation is to evaluate the safety and effectiveness of the Navitor TAVI System for treating patients with symptomatic, severe native aortic stenosis who are considered intermediate or low risk for surgical mortality. Subjects in this clinical investigation will be randomized between the Navitor TAVI System (test arm) and any commercially available TAVI system (control arm) in a 1:1 ratio.
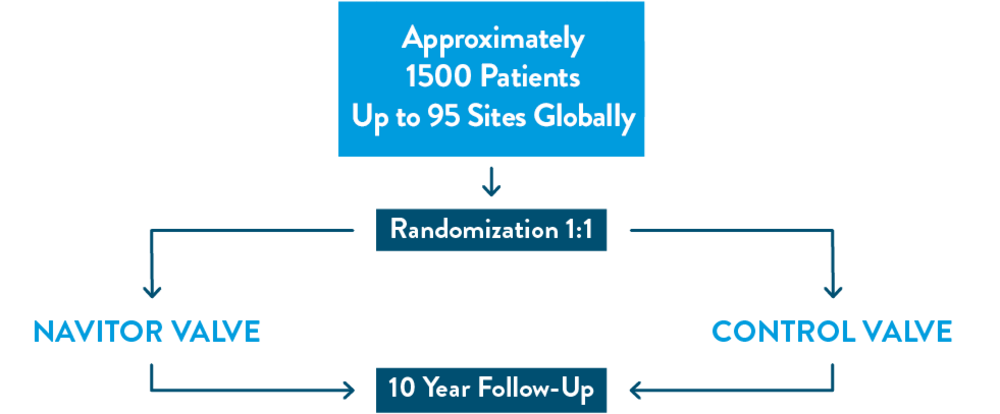
- Clinical trial endpoints
Primary endpoint
Composite of all-cause mortality or all stroke at 12 months
Find a clinical trial site near me

The REPAIR MR Trial (Percutaneous MitraClip™ Device or Surgical Mitral Valve REpair in PAtients with PrImaRy Mitral Regurgitation who are Candidates for Surgery) is now underway, and will be studying 500 patients at up to 75 medical centers in the United States, Canada, and Europe.
This clinical trial will study the MitraClip™ Transcatheter Mitral Valve Repair system in patients with severe primary mitral regurgitation who are at moderate surgical risk and whose mitral valve (MV) has been determined to be suitable for correction by MV repair surgery by the cardiac surgeon on the local site heart team. The objective of this trial is to compare the clinical outcomes of the MitraClip device versus surgical repair in patients with severe primary MR who are at moderate surgical risk.
CAUTION: MitraClip (for REPAIR MR indications) is an investigational device. Limited by Federal (U.S.) law to investigational use only.
- Procedural overview
Transcatheter mitral valve repair is a less-invasive treatment option for patients who are deemed appropriate for this procedure. The procedure is done using the MitraClip Transcatheter Mitral Valve Repair system.

1. A guide catheter (a thin tube) will be inserted through a vein from a small cut in your upper leg to reach your heart.

2. The MitraClip implant, which is attached to the end of a clip delivery system, will be guided to your mitral valve through the catheter.

3. The implant will be positioned and the clip will grasp the mitral valve leaflets to close the center of the mitral valve and reduce MR.

4. Once the clip is in place, it will be separated from the clip delivery system.

5. The implanted clip will become a permanent part of your heart, allowing your mitral valve to close more tightly and reduce the backward flow of blood.
- Eligibility criteria*
Key inclusion criteria
Subjects must meet all of the following inclusion criteria in order to participate in the trial:
- Subject has severe (Grade III or greater per the ASE criteria, which includes severity grades of 3+ and 4+) primary MR as assessed by the echocardiography core-lab (mixed etiology is acceptable provided the principal mechanism of action is due to degenerative mitral valve pathology)
- The cardiac surgeon of the site Heart Team (consisting of at least one interventionalist, and one cardiac surgeon) has confirmed that the subject is a candidate for mitral valve surgery and the eligibility committee (EC) has confirmed that the subject’s mitral valve anatomy is suitable for percutaneous repair with the MitraClip™ device with high certainty of achieving MR ≤ mild
- Subject is symptomatic (NYHA Class II/III/IV) or asymptomatic with LVEF ≤ 60%, pulmonary artery systolic pressure > 50 mmHg, or LVESD > 40 mm
- Subject is at least 75 years of age, OR if younger than 75 years, then has:
- Society of Thoracic Surgeons (STS) Predicted Risk of Mortality (PROM) Repair Score ≥ 2%, or
- Presence of other comorbidities which may introduce a potential surgery-specific impediment
- Subject provides written informed consent
- Subject is ≥ 18 years of age
Key exclusion criteria
Subject must not meet any of the following exclusion criteria in order to participate in the trial:
- Subject is currently participating in another clinical investigation
- Presence of other anatomic or comorbid conditions, or other medical, social, or psychological conditions that, in the investigator’s opinion, could limit the subject’s ability to participate in the clinical investigation or to comply with follow-up requirements, or impact the scientific soundness of the clinical investigation results.
- Subject has ischemic or non-ischemic secondary MR
- Concomitant severe tricuspid valve regurgitation
- Ejection fraction < 30%
- Severe mitral annular calcification
- Acute myocardial infarction in the past 12 weeks
- Need for cardiac surgery to correct pulmonary valve disease, aortic valve disease, or tricuspid valve disease
- Subjects who have concurrent coronary artery disease may be included provided the subjects are eligible for both percutaneous coronary intervention (PCI) and coronary artery bypass surgery. Subjects randomized to the device group, must undergo PCI before the MitraClip procedure. Subjects randomized to the surgical (control) arm may undergo coronary artery revascularization during mitral valve repair surgery.
- Surgical procedure performed in the past 30 days
- Femoral vein cannot accommodate a 24 F catheter or presence of IVC filter would interfere with the catheter or ipsilateral DVT
- Transesophageal echocardiography (TEE) is contraindicated
- Hemodynamic instability: systolic pressure ≤ 90 mmHg without afterload reduction, cardiogenic shock, or the need for inotropic support or Intra-Aortic Balloon Pump (IABP)
- Need for emergency surgery for any reason
- Prior mitral valve surgery, valvuloplasty, mechanical prosthetic valve or Ventricular Assist Device (VAD)
- Systolic anterior motion of the Mitral Valve
- Hypertrophic cardiomyopathy
- Renal insufficiency requiring dialysis
- Active infections requiring current antibiotic therapy
- Subjects who are pregnant or planning to be pregnant
*Additional inclusion and exclusion criteria as listed on ClinicalTrials.gov - Trial design
This is a prospective, multi-center, randomized, controlled, clinical investigation of 500 subjects at 60 medical centers in the United States, Canada, and Europe.

Clinical follow-ups will be a combination of in-person and phone only follow ups.
Between year 2 and 10 follow ups will take place every year.

Clinical follow-ups will be a combination of in-person and phone only follow ups.
Between year 2 and 10 follow ups will take place every year.
- Clinical trial endpoints
Co-primary endpoint #1
All-cause mortality, stroke, cardiac hospitalization, or acute kidney injury requiring renal replacement therapy at 2 years (any cardiac hospitalizations in the first 30 days post treatment will be excluded)
Co-primary endpoint #2
Proportion of subjects with moderate or less MR (≤2+), without mitral valve replacement, and without recurrent mitral valve intervention (surgical or percutaneous) from the time of index procedure through 2 years
Secondary endpoints
The following secondary endpoints will be assessed and compared between device and control:
- Proportion of subjects with MR ≤ mild (1+) at 30 days post-index procedure among survivors
- Hospital length of stay from procedure to home discharge (days)
- Proportion of subjects discharged to home post index hospitalization
- Quality of life improvement assessed using the Kansas City Cardiomyopathy Questionnaire (KCCQ) of at least 10 points at 2 years compared to baseline among survivors.
- Severe symptomatic mitral stenosis at 1 year
*TMVr is also referred to as TEER (transcatheter edge-to-edge repair)
Find a clinical trial site near me

SUMMIT trial examines transcatheter mitral valve replacement (TMVR) with Tendyne™ mitral valve
The SUMMIT Trial (Clinical Trial to Evaluate the Safety and Effectiveness of Using the Tendyne Transcatheter Mitral Valve System for the Treatment of Symptomatic Mitral Regurgitation) is now underway, and will be enrolling patients at up to 80 sites in the U.S., Canada, Europe, and Japan.1
The trial is examining a treatment with the Tendyne Transcatheter Mitral Valve System, for patients who have symptomatic, moderate-to-severe or severe mitral regurgitation (MR) or mitral annular calcification (MAC) and who are appropriate candidates for TMVR. The objective of this trial is to:
- Determine whether this technology improves clinical outcomes for the large number of patients suffering from the debilitating symptoms of moderate-to-severe or severe MR
- Compare the TMVR outcomes to MitraClip™ transcatheter mitral valve repair, the standard of care for patients not suitable for mitral valve surgery.
CAUTION: Tendyne is an investigational device. Limited by Federal (U.S.) law to investigational use only.
- Procedural overview
The Tendyne Transcatheter Mitral Valve is a bioprosthesis designed to reduce MR and improve blood flow through the heart. The following steps provide a general overview of the Tendyne procedure.

1. Access to the heart is gained through a small incision in the chest between the ribs.

2. The Tendyne valve is placed in the heart through a catheter.
3. The tether is secured to a pad at the bottom of the heart. The valve, tether, and pad are designed to become a permanant implant, reduce MR, and improve blood flow. - Eligibility criteria*
Key Inclusion Criteria
- Symptomatic, moderate-to-severe or severe mitral regurgitation, or severe mitral annular calcification (MAC)
- NYHA Functional Classification ≥ II (if Class IV, patient must be ambulatory)
- The local site heart team determines that the subject has been adequately treated per applicable standards
- Not a member of a vulnerable population
Key Exclusion Criteria
- Mitral valvular vegetation or mass
- Left ventricular ejection fraction < 25%
- Left ventricular end diastolic diameter > 7.0 cm
- Prior surgical or interventional treatment of mitral valve involving implantation of prosthetic material
- Aortic valve disease requiring surgery or transcatheter intervention
- Severe tricuspid regurgitation or any tricuspid valve disease requiring surgery or transcatheter intervention
- Any planned surgical / interventional procedure within 60 day prior to or following subject randomization
- Subject undergoing hemodialysis due to chronic renal failure
- Mitral pathoanatomy and left ventricular outflow tract anatomy deemed not suitable for Trial device implantation
- Subjects with non-cardiac comorbidities that are likely to result in a life expectancy of less than 12 months
*Additional inclusion and exclusion criteria as listed on ClinicalTrials.gov - Trial design
This is a prospective, controlled, multi-center clinical investigation. Investigators anticipate a 48-month recruitment period, with subject follow-up continuing for an additional 60 months.

*2017 ASE Guidelines.
†Associated with MR ≥ Grade III, severe MS, or moderate MR with moderate MS.
*2017 ASE Guidelines.
†Associated with MR ≥ Grade III, severe MS, or moderate MR with moderate MS. - Clinical trial endpoints
Randomized cohort (vs. MitraClip)
Survival free from heart failure hospitalization at 12 months post index procedure
Non-repairable cohort
Freedom for death or HFH
MAC cohort
Survival free from heart failure hospitalization at 12 months post index procedure
All cohorts
The following additional endpoints will be assessed and compared between device and control:
- MVARC-defined Technical, Device, Procedural, and Patient Success
- Annualized rate of heart failure hospitalization
- All strokes, or mitral valve reintervention/reoperation

MVARC = Mitral Valve Academic Research Consortium.
To learn more about the SUMMIT Trial, including eligibility criteria and study locations, please visit the ClinicalTrials.gov website.

Landmark study of a minimally invasive device for patients with severe tricuspid regurgitation
The TRILUMINATE™ Pivotal Trial (Clinical Trial to Evaluate Cardiovascular Outcomes In Patients Treated With the Tricuspid Valve Repair System) is a prospective, randomized, controlled trial designed to compare the TriClip™ TEER System to Medical Therapy in symptomatic patients with severe tricuspid regurgitation (TR).
The objective of this trial is to evaluate the safety and effectiveness of the TriClip™ TEER System in improving clinical outcomes in symptomatic patients with severe TR who have been determined by the site local heart team to be at intermediate or greater estimated risk for mortality with tricuspid valve surgery.
TriClip™ TEER is a clip-based tricuspid transcatheter edge-to-edge repair system, delivered via a minimally invasive procedure with no need for cardiopulmonary bypass or arresting the heart.
CAUTION: The TriClip TEER System is an investigational device. Limited by Federal (U.S.) law to investigational use only.
- Procedural overview
TriClip TEER is a minimally invasive transcatheter procedure performed on a beating heart in a standard cath lab or hybrid room. The TriClip™ Delivery System is anatomically designed for direct access to the tricuspid valve via the right atrium. The following steps provide a general overview of the TriClip TEER procedure.
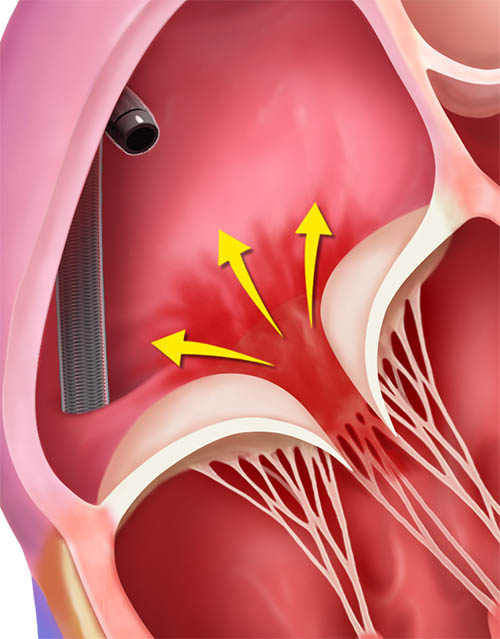
TriClip™ Steerable Guide Catheter insertion into the right atrium
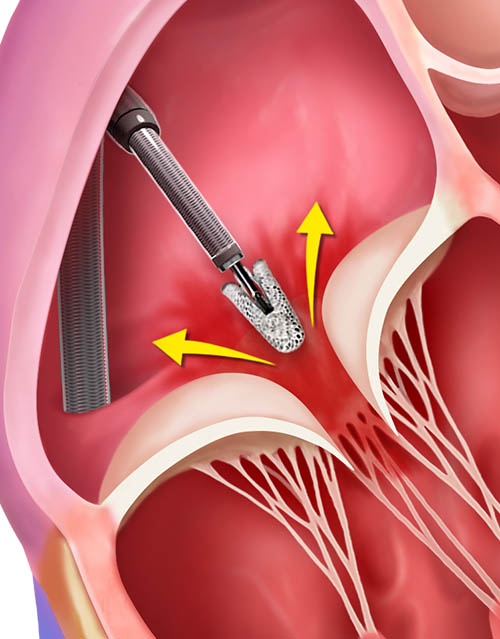
TriClip Delivery System insertion, positioning and steering in the right atrium
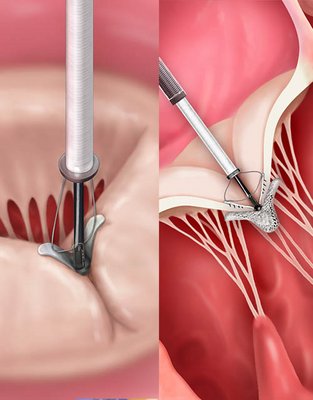
Crossing the tricuspid valve and grasping leaflets
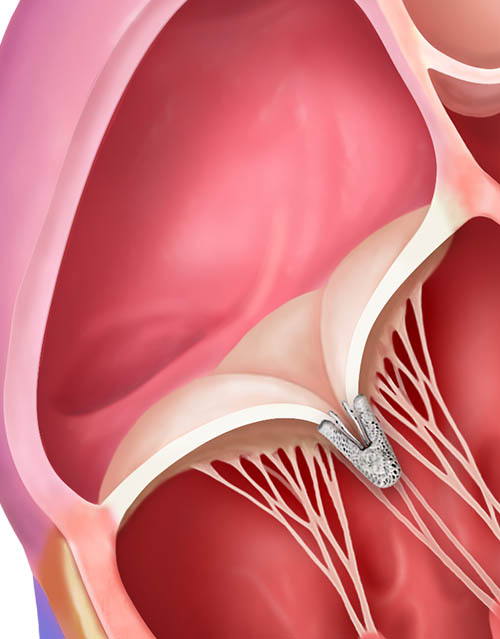
TriClip™ Implant deployed, delivery system removed
- Eligibility criteria*
Key inclusion criteria
Subjects must meet all of the following inclusion criteria in order to participate in the trial:
- In the judgment of the site’s local heart team, subject has been adequately treated per applicable standards (including medical management) and stable for at least 30 days as follows:
- Optimized medical therapy for treatment of TR (eg, diuretics)
- Medical and/or device therapy for mitral regurgitation, atrial fibrillation, coronary artery disease, and heart failure
- The eligibility committee (EC) will confirm that the subject has been adequately treated medically
- Subject is symptomatic with severe TR despite being optimally treated as described above. TR severity is determined by the assessment of a qualifying transthoracic echocardiogram (TTE) and confirmed by the Echocardiography Core Lab (ECL). The ECL may request a transesophageal echocardiogram (TEE) to confirm TR etiology. Note: If any cardiac procedure(s) occur after eligibility was determined, TR severity will need to be re-assessed 30 days after the cardiac procedure(s)
- The cardiac surgeon of the Site Heart Team concurs that the patient is at intermediate or greater estimated risk of mortality with tricuspid valve surgery
- New York Heart Association (NYHA) Functional Class II, III, or ambulatory class IV
Key exclusion criteria- Systolic pulmonary artery pressure (sPAP) > 70 mm Hg or fixed pre-capillary pulmonary hypertension as assessed by right heart catheterization (RHC)
- Severe uncontrolled hypertension (systolic blood pressure [SBP] ≥ 180 mm Hg and/or diastolic blood pressure [DBP] ≥ 110 mm Hg)
- Indication for left-sided (eg, severe aortic stenosis, severe mitral regurgitation) or pulmonary valve correction in the prior 60 days
- Left ventricular ejection fraction (LVEF) ≤ 20%
- Pacemaker or implantable cardioverter defibrillator (ICD) leads that would prevent appropriate placement of the TriClip™ device
- Any prior tricuspid valve procedure that would interfere with placement of the TriClip™ device
- Tricuspid valve leaflet anatomy which may preclude clip implantation, proper clip positioning on the leaflets, or sufficient reduction in TR. This may include:
- Evidence of calcification in the grasping area
- Presence of a severe coaptation defect (> 2 cm) of the tricuspid leaflets
- Severe leaflet defect(s) preventing proper device placement
- Ebstein anomaly
- Tricuspid valve stenosis (tricuspid valve orifice of ≤ 1.0 cm2 and/or mean gradient ≥ 5 mm Hg as measured by the ECL)
- Tricuspid valve anatomy not evaluable by TTE and TEE
*Additional inclusion and exclusion criteria as listed on ClinicalTrials.gov - In the judgment of the site’s local heart team, subject has been adequately treated per applicable standards (including medical management) and stable for at least 30 days as follows:
- Trial design
TRILUMINATE is a prospective, randomized, controlled, multicenter trial that is enrolling up to 450 subjects.

- Clinical trial endpoints
Primary Endpoint:
To be assessed after the first 350 randomized subjects complete 12-month follow-up.
A composite of mortality or tricuspid valve surgery, heart failure hospitalizations, and quality of life improvement ≥15 points assessed using the Kansas City Cardiomyopathy Questionnaire (KCCQ), evaluated at 12 months in a hierarchical fashion using the Finkelstein-Schoenfeld methodology.
Secondary Endpoints:Assessed hierarchically in the following order:
- Freedom from major adverse events (MAE) after procedure attempt (femoral vein puncture) at 30 days (Device group only)
- Change in KCCQ at 12 months (superiority of Device vs. Control)
- TR Reduction to moderate or less at 30-day post procedure (superiority of Device vs. Control)
- Change in 6MWD at 12 months (superiority of Device vs. Control)
To learn more about the TRILUMINATE Pivotal Trial, including eligibility criteria and study locations, please visit the ClinicalTrials.gov website.
MAT-2115170 v10.0 Item approved for U.S. use only.